The delicate balance of bone remodeling is essential for maintaining skeletal strength and integrity. Among the various regulators of this process, cortisol plays a pivotal role. While it is critical for the body’s stress response and metabolic regulation, excessive cortisol levels can disrupt the harmony between bone formation and resorption. This article delves into the multifaceted ways in which cortisol influences bone biology, examines the clinical implications of chronic hypercortisolemia, and explores strategies to protect the skeleton from glucocorticoid-induced damage.
Mechanisms of Cortisol in Bone Biology
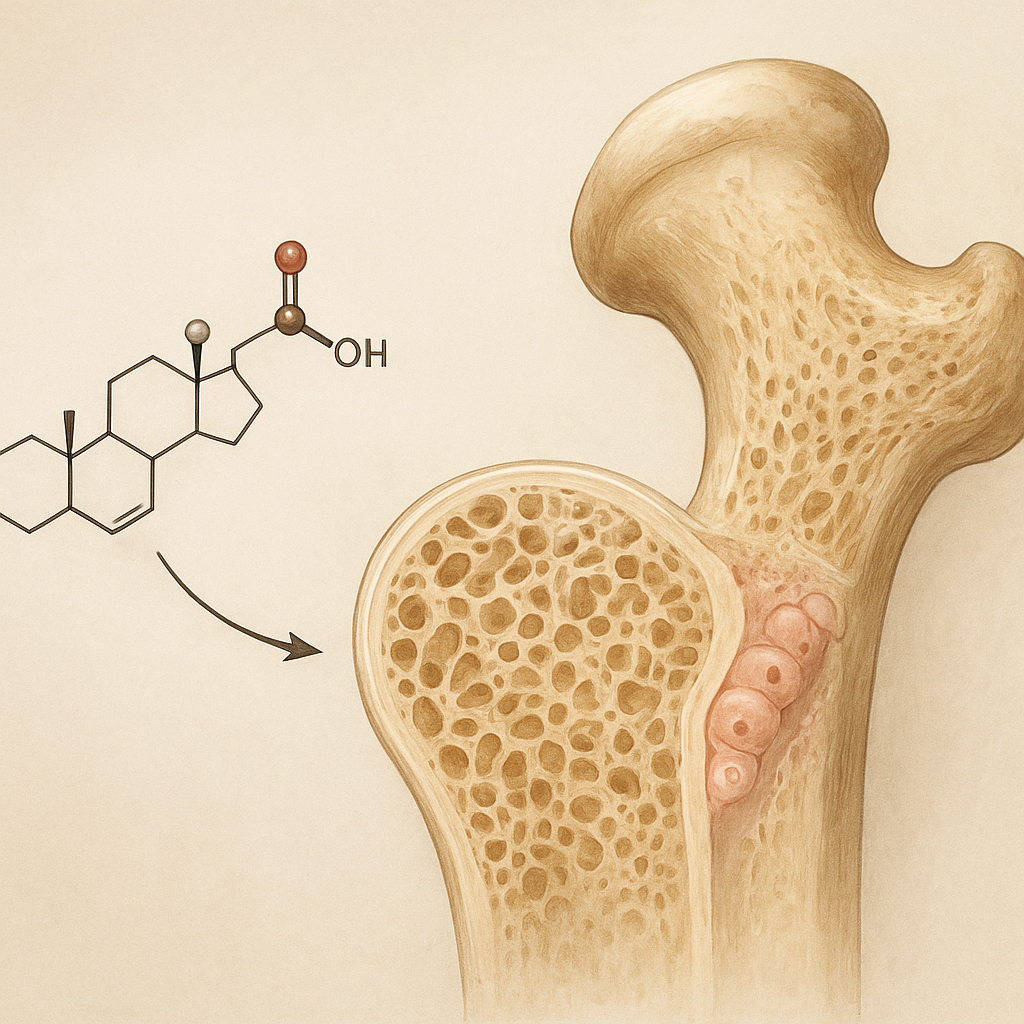
Cortisol, a primary glucocorticoid hormone synthesized by the adrenal cortex, exerts its effects via binding to intracellular receptors that modulate gene expression. In the context of bone, two cell populations—osteoblasts (bone-forming cells) and osteoclasts (bone-resorbing cells)—are the main targets.
Glucocorticoid Receptors and Osteoblast Function
Osteoblasts express both glucocorticoid receptor alpha (GRα) and beta (GRβ) isoforms. Upon cortisol binding, GRα translocates to the nucleus, where it influences transcription of genes involved in:
- Matrix protein production, including type I collagen synthesis
- Growth factors such as insulin-like growth factor (IGF-1)
- Cellular proliferation and differentiation
Chronic elevation of cortisol induces apoptosis in osteoblasts, reduces the expression of osteogenic factors, and downregulates RUNX2, a master transcription factor critical for osteoblast lineage commitment. The net effect is diminished bone formation and impaired mineral deposition.
Impact on Osteoclast Activity
Although cortisol suppresses osteoblasts, its influence on osteoclasts is more complex. Under normal conditions, glucocorticoids inhibit inflammatory cytokines, indirectly reducing osteoclastogenesis. However, prolonged exposure leads to:
- Increased production of receptor activator of nuclear factor kappa-B ligand (RANKL) by stromal cells
- Reduced secretion of osteoprotegerin (OPG), a decoy receptor for RANKL
- Enhanced survival and activity of mature osteoclasts
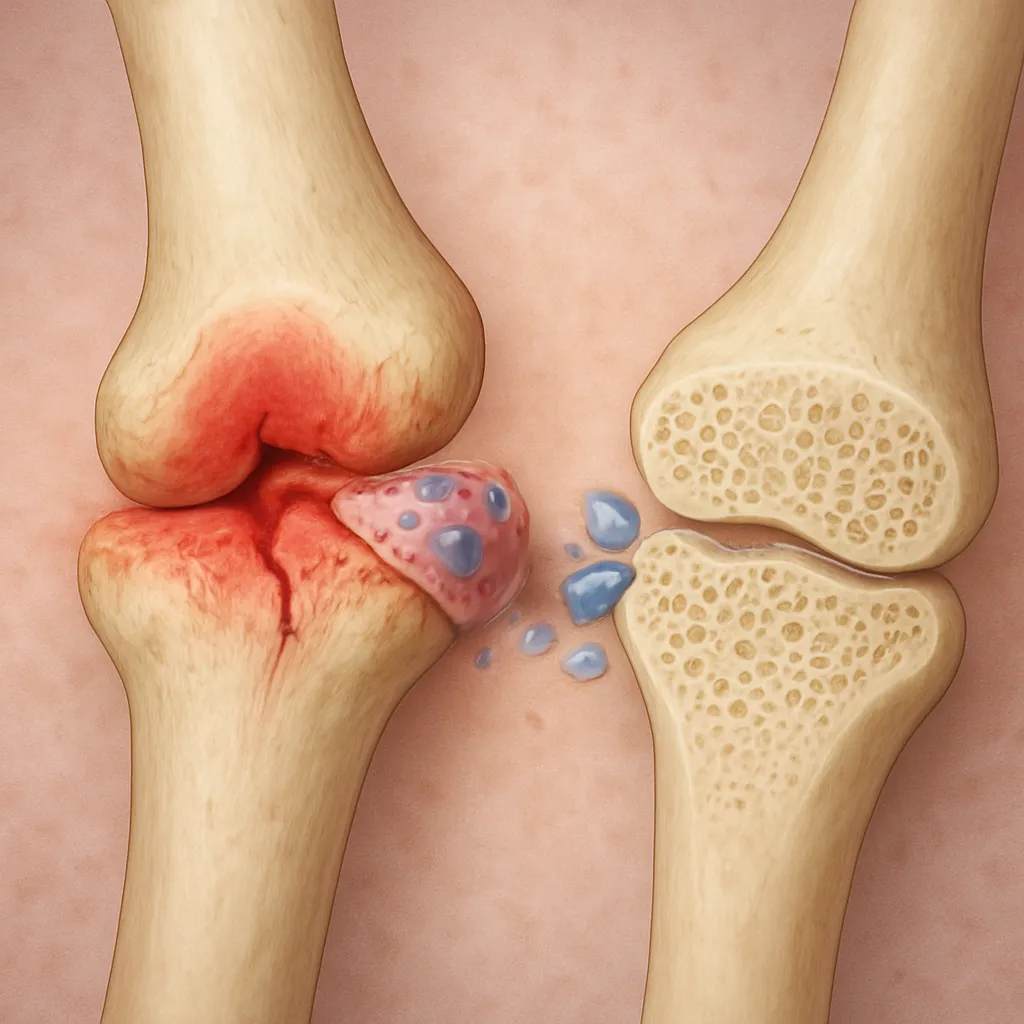
The combined effect skews the remodeling balance toward excess bone resorption, weakening the trabecular architecture and compromising bone mineral density (BMD).
Chronic Stress, Cortisol, and Skeletal Health
Persistent stress stimulates the hypothalamic-pituitary-adrenal (HPA axis), leading to sustained cortisol release. The resulting hypercortisolemia can have devasting consequences on the skeleton, particularly when exacerbated by other risk factors such as aging, nutritional deficiencies, or genetic predispositions.
Stress Response and Bone Remodeling
Acute stress triggers a transient spike in cortisol that mobilizes energy reserves and modulates immune responses. In contrast, chronic stress leads to:
- Downregulation of bone anabolic signals
- Elevation of pro-resorptive mediators (e.g., interleukin-6)
- Suppression of sex hormones (estrogen, testosterone), which are protective to bone
This milieu fosters microarchitectural deterioration, particularly in trabecular-rich sites such as the lumbar spine and proximal femur.
Clinical Conditions: Cushing’s Syndrome and Exogenous Glucocorticoids
Cushing’s syndrome—characterized by endogenous cortisol overproduction—serves as a natural model for glucocorticoid-induced osteoporosis. Patients often present with:
- Vertebral compression fractures
- Proximal muscle weakness
- Delayed fracture healing
Similarly, long-term therapy with synthetic glucocorticoids (prednisone, dexamethasone) for autoimmune disorders or transplant rejection poses a significant risk to bone health. Clinical studies reveal a rapid decline in bone mineral density within the first six months of high-dose therapy, followed by a more gradual but continual loss.
Therapeutic Strategies to Mitigate Cortisol-Induced Bone Loss
Given the essential roles of glucocorticoids in medicine, complete avoidance is often impractical. Instead, strategies focus on minimizing skeletal side effects while preserving therapeutic efficacy.
Pharmacological Interventions
- Bisphosphonates: Inhibit osteoclast-mediated resorption and have proven efficacy in preventing fractures in glucocorticoid-treated patients.
- Denosumab: A monoclonal antibody against RANKL, effective in reducing bone turnover and increasing BMD.
- Teriparatide: Recombinant parathyroid hormone (PTH 1-34) that stimulates osteoblast activity and improves trabecular microarchitecture.
- Selective estrogen receptor modulators (SERMs): Provide estrogenic protection to bone without adverse effects on breast and uterine tissues.
Regular monitoring of BMD and bone turnover markers guides the choice and duration of these agents.
Nutrition, Lifestyle, and Alternative Approaches
Complementary measures can bolster skeletal resilience against cortisol’s catabolic effects:
- Optimal intake of calcium and vitamin D to support mineralization and parathyroid hormone regulation.
- Weight-bearing and resistance exercise to stimulate mechanotransduction pathways in osteocytes.
- Stress management techniques (mindfulness, cognitive behavioral therapy) to attenuate HPA axis hyperactivity.
- Limiting alcohol consumption and tobacco use, both of which exacerbate bone loss.
Emerging research on selective glucocorticoid receptor modulators aims to retain anti-inflammatory benefits while sparing bone tissue, offering hope for future therapies.